

A force field for energy calculations and protein design
From CRG - EMBL -VIB
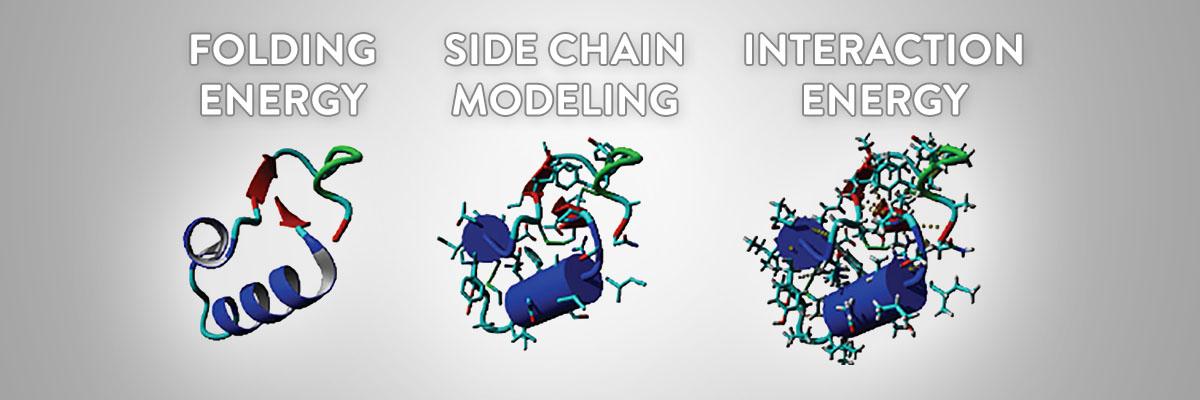
Based on FoldX
FoldX combines a physical description of the interactions with lessons learned from experiments.

FoldX Suite: Modular and portable
LoopX and PepX are fully integrated with FoldX

LoopX
Protein Loop Structure Prediction Algorithm

PepX
Peptide Docking Algorithm
The FoldX Suite is available through academic and commercial licenses. Click on the boxes to obtain more information about them. We also offer Consulting Services tailored to our customer’s needs.
The FoldX Suite builds on the strong fundament of advanced protein design features, already implemented in the successful FoldX3, and exploits the power of fragment libraries, by integrating in silico digested backbone protein fragments of different lengths. Such fragment-based strategy allows for new powerful capabilities: loop reconstruction, implemented in LoopX and peptide docking, implemented in PepX. The Suite also features an improved usability, thanks to a new boost Command Line Interface.